
Other commentaries
HSA/hFcRn, a powerful model for your PK/PD studies
6 min read
July 14, 2020
Read detailed information on the HSA/hFcRn mouse model
The old adage "Begin with the end in mind" is particularly appropriate for preclinical drug development because the choice of a preclinical model is key to support the intended clinical trial design. For the past 20 years, the chances of success for a compound entering phase I trials have been slightly under 10%, which means that for every 20 compounds reaching clinical trials, only two have obtained clinical use authorization. Notably, in 75–80% of cases, these failures appear to relate to the inability of drugs found efficacious in animal models to translate to clinical trials, leading some researchers to call this translational gap the “Valley of Death”.1,2 It is indeed difficult to find an animal model that not only delineates the safety and toxicity of a potential drug candidate but accurately predicts its dose and efficacy, ultimately supporting the intended clinical trial design.3
Another challenge is to select the best formulation of a drug to target precise regions of the body, and maximize the quantity and duration of its exposure. As therapeutics are often low-molecular-weight molecules, they have rapid renal clearance, short plasma circulatory time, and nonspecific distribution, all contributing to a weak, short and diffuse therapeutic action.3 To overcome these obstacles, tremendous efforts have been made to generate drug delivery systems, i.e., engineered technologies for the targeted delivery and/or controlled release of therapeutic agents.
One example of such technologies is represented by human serum albumin (HSA), the most abundant protein in the blood (60% by mass) that, over the past decades, has become a pivotal contender as a molecular cargo and nano vehicle in biophysical, clinical and industrial fields due to its extraordinary capacity to bind, store and transport a wide variety of endogenous and exogenous ligands, including pharmacological drugs.4
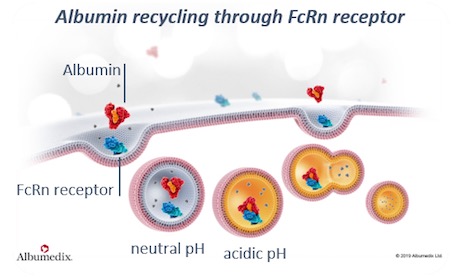
HSA's long circulatory half-life (~19 days) is favored by interaction with the neonatal Fc receptor (FcRn), a quasi-ubiquitous heterodimeric protein consisting of a transmembrane alpha-heavy chain noncovalently associated with a beta-2-microglobulin light chain.5,6 FcRn facilitates the transport of maternal IgG to the growing fetus, prevents the intracellular catabolism of albumin and IgG, and transports these two ligands bidirectionally across cellular membranes.5,7,8Interestingly, recombinant human albumins engineered for enhanced FcRn binding display protracted blood circulation profiles in mice, thereby representing excellent tools to deliver potential drugs at the target site, at a concentration and frequency that maximize its therapeutic action.9
Mice serve an important role in preclinical drug development, as they help to delineate the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of new potential therapeutic agents.3 Historically, two mouse models have been widely used to assess the PK properties of HSA-based drugs, Tg32 and Tg276; however, due to cross-species differences in the albumin/FcRn interaction, the usefulness of these models has been questioned in PK studies of HSA-based therapeutics. A study conducted by Jan Terje Andersen and colleagues revealed that murine serum albumin (MSA) displays a higher affinity for hFcRn than HSA, and competes with the latter by inhibiting receptor binding.10 Both Tg32 and Tg276 mice express the endogenous MSA but not the exogenous HSA, which almost certainly affects PK analyses of HSA-based compounds.10,11 Furthermore, both mice express the hFcRn alpha chain either under the control of the native human promoter (for Tg32) or the ubiquitous CAG promoter (for Tg276), so the absence of an endogenous mouse promoter-driven FcRn expression could lead to a nonnative distribution.6
To overcome the limits of these two models, a group of researchers led by Kenneth A. Howard generated, in collaboration with genOway, a double-humanized SA and FcRn alpha chain (HSA/hFcRn) mouse model that displays physiological expression level of HSA (within the normal range of 1.5–6 g/dL), and normal blood chemistry.6 Moreover, since both HSA and hFcRn are controlled by their respective endogenous mouse promoters, and the expression of MSA and mFcRn is abolished, this mouse represents a physiologically relevant model for the preclinical study of albumin-based drugs, conventional drugs, and biologics whose action is influenced by reversible binding to endogenous HSA.
Although the “Valley of Death” is far from bridged, optimized animal models such as HSA/hFcRn certainly contribute to narrowing the translational gap. In this spirit, and to widen the fields of application of the model, the HSA/hFcRn mouse is now also available on the immunodeficient background Rag1-null. It can therefore be directly used in xenograft studies, and to assess the therapeutic efficacy of drugs in primary immunodeficiency diseases, immuno-oncology, and infectious diseases.
See also:
Part 1: Toward a Better Effective Preclinical Model in Immuno-Oncology
Part 2: BRGSF, a New Immunodeficient Model for Immuno-Oncology Studies
Part 3: BRGSF-HIS, a New Human Immune System Mouse Model for Immuno-Oncology Studies
Part 4: Humanized immune checkpoint mouse models for immuno-oncology studies
References:
- Dowden H, Munro J. Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov. 2019;18(7):495-496. doi:10.1038/d41573-019-00074-z
- Butler D. Translational research: Crossing the valley of death. Nature. 2008;453(7197):840-842. doi:10.1038/453840a
- Steinmetz KL, Spack EG. The basics of preclinical drug development for neurodegenerative disease indications. BMC Neurol. 2009;9(Suppl 1):S2. doi:10.1186/1471-2377-9-S1-S2
- Al-Harthi S, Lachowicz JI, Nowakowski ME, Jaremko M, Jaremko Ł. Towards the functional high-resolution coordination chemistry of blood plasma human serum albumin. Journal of Inorganic Biochemistry. 2019;198:110716. doi:10.1016/j.jinorgbio.2019.110716
- Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The Neonatal Fc Receptor (FcRn): A Misnomer? Front Immunol. 2019;10:1540. doi:10.3389/fimmu.2019.01540
- Viuff D, Antunes F, Evans L, et al. Generation of a double transgenic humanized neonatal Fc receptor (FcRn)/albumin mouse to study the pharmacokinetics of albumin-linked drugs. Journal of Controlled Release. 2016;223:22-30. doi:10.1016/j.jconrel.2015.12.019
- Brambell FWR. THE PASSIVE IMMUNITY OF THE YOUNG MAMMAL. Biological Reviews. 1958;33(4):488-531. doi:10.1111/j.1469-185X.1958.tb01412.x
- Chaudhury C, Mehnaz S, Robinson JM, et al. The Major Histocompatibility Complex–related Fc Receptor for IgG (FcRn) Binds Albumin and Prolongs Its Lifespan. Journal of Experimental Medicine. 2003;197(3):315-322. doi:10.1084/jem.20021829
- Andersen JT, Dalhus B, Cameron J, et al. Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat Commun. 2012;3(1):610. doi:10.1038/ncomms1607
- Terje Andersen J, Bekele Daba M, Berntzen G, Michaelsen TE, Sandlie I. Cross-species Binding Analyses of Mouse and Human Neonatal Fc Receptor Show Dramatic Differences in Immunoglobulin G and Albumin Binding. J Biol Chem. 2010;285(7):4826-4836. doi:10.1074/jbc.M109.081828
- Proetzel G, Roopenian DC. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods. 2014;65(1):148-153. doi:10.1016/j.ymeth.2013.07.005
Related products
Catalogue product
Customized product
No items found.
Scientific excellence
From model design to experimental results
Featured in 600+ scientific articles
Collaborative approach
Collaboration with 17 Top Pharmas,
170+ Biotechs and 380+ Academic Institutions
Robust validation data on catalog models
Generated with biopharma partners and in-house
Innovative technologies
and guaranteed freedom to operate
Easy access to models
Models with certified health status from professional breeders in US and Europe